Zweck der Prüfung und Rückverfolgbarkeit
Der Zweck der Prüfung besteht darin, sicherzustellen, dass ein Prozess oder System konsistent ist und dokumentiert wird. Die Systemprüfung ist eine Anforderung der Regulierungsbehörden. Zu den Regulierungsbehörden für Life-Science-Organisationen gehört beispielsweise die US-amerikanische Food and Drug Administration (FDA).
Die FDA definiert Prüfung wie folgt:
Bestätigung durch Prüfung und Bereitstellung objektiver Nachweise, dass die besonderen Anforderungen für einen bestimmten Verwendungszweck durchgängig erfüllt werden können.
Die Weltgesundheitsorganisation (WHO) definiert Prüfung entsprechend ihren Leitlinien zu den Anforderungen an eine gute Herstellungspraxis (Good Manufacturing Practice, GMP) wie folgt:
Erbringung eines dokumentarischen Nachweises, der ein hohes Maß an Sicherheit bietet, dass ein geplanter Prozess einheitlich mit den erwarteten spezifizierten Ergebnissen übereinstimmt.
Diese Definitionen haben gemäß den erwarteten Ergebnissen die folgenden Elemente gemeinsam:
- Generierung von Beweisen
- Einhaltung der Vorschriften
- Erfüllung der Anforderungen
Die Prüfung computergestützter Systeme ist ein dokumentierter Prozess, der sicherstellt, dass das System auf konsistente und reproduzierbare Weise seinen Zweck erfüllt. Die Prüfung stellt die Integrität und Sicherheit der Datenverarbeitung, die Produktqualität und die Einhaltung von Vorschriften sicher, die für Good {industry} Practice (GxP) gelten.
Die Vorgehensweise bei der Prüfung eines computergestützten Systems wird in den Standardarbeitsanweisungen (Standard Operating Procedure, SOP) und Richtlinien beschrieben, die von der regulierten Industrie wie z. B. Life Science-Organisationen erstellt und definiert werden. Für die Prüfung computergestützter Systeme ist es sinnvoll, die Umsetzung des Prozesses als ein Projekt zu betrachten, siehe Good Automated Manufacturing Practice (GAMP) 5: Ein risikobasierter Ansatz für konforme computergestützte GxP-Systeme der International Society for Pharmaceutical Engineering's (ISPE's).
Bevor Sie mit dem Implementierungsprojekt beginnen, sollte ein allgemeiner Plan für die neue Lösung vorliegen. Starten Sie dann das Projekt, indem Sie die folgenden Phasen abschließen:
- Planung: In dieser Phase sollten die Anforderungen und Spezifikationen klar genug sein für eine erste Risikobewertung und letztendlich für eine korrekte Definition von Verifizierungstests (Protokolle). Während dieser Phase stellen Sie das Dokument für den Prüfungsplan bereit, das die gesamte Prüfungsstrategie und die zu erbringenden Leistungen definiert. Die Strategie sollte mit dem Quality Management System (QMS) und den Richtlinien übereinstimmen.
- Spezifikation, Konfiguration und Codierung: In dieser Phase werden alle Designspezifikationen mit der Detailebene erstellt, die für den Systemtyp und seiner Verwendung erforderlich ist. Die Entwickler*innen wählen die für die Codierungs- und Konfigurationsanforderungen am besten geeigneten und auf den genehmigten Spezifikationen basierenden Entwicklungsmethoden und -Objekte aus und verwenden diese. Alle diese Aktivitäten werden in der Entwicklungsumgebung durchgeführt. In dieser Phase konzentriert sich das Testen mehr auf die Überprüfung der Einheiten oder Funktionen aus Entwicklungssicht. Beispiele für solche Tests sind Komponententests, statistische Codetests und Integrationstests. Tools können diese Testaktivitäten automatisieren.
- Testen: In dieser Phase wird bestätigt, dass die Spezifikationen durch Inspektionen und Tests des Systems erfüllt wurden. Die Testaktivitäten werden in einer vorbereiteten und geeigneten Testumgebung durchgeführt. Die Testumgebung muss der Produktionsumgebung ähneln, um sicherzustellen, dass die Bedingungen gleich sind und Sie die Tests in der Produktionsumgebung nicht wiederholen müssen. Das Risiko sollte den Umfang des Testaufwands steuern. Die Risikoanalyse kann Ihnen dabei helfen, potenzielle Gefahren zu verstehen, die sich auf die Produktqualität, die Patientensicherheit oder die Datenintegrität auswirken können. Diese potenziellen Gefahren müssen durch Kontrollen und Nachweise von Tests gemindert werden. Wenn irgendwo ein hohes Risiko besteht, sollten Sie über geeignete Testszenarien verfügen, um nachzuweisen, dass der Lösungsentwurf ohne Fehlerpotenzial ist.
- Berichterstellung und Freigabe: In dieser Phase muss das System gemäß einem dokumentierten und kontrollierten Prozess für den Einsatz in der Produktionsumgebung akzeptabel sein. Bei Projektabschluss muss ein Abschlussbericht über die Systemprüfung erstellt werden, in dem die durchgeführten Aktivitäten und eventuelle Abweichungen vom Prüfungsplan zusammengefasst werden. Die Prüfung des Systems sollte vor der Freigabe zur Nutzung abgeschlossen sein.
Die folgende Abbildung zeigt das V-Modell, das von GAMP 5, 2. Edition unterstützt wird. Es bietet eine gute Übersicht über die Projektphasen.
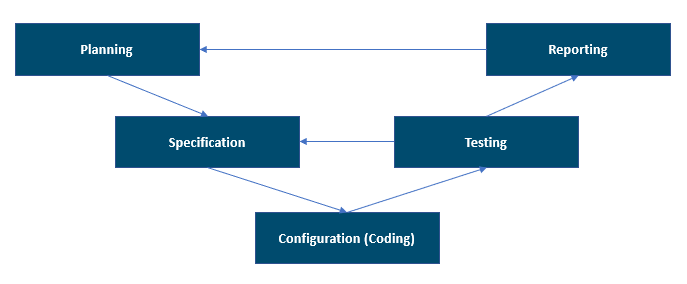
Das V-Modell kann nicht nur als die Entwicklungs- und Testaktivitäten des Systems betrachtet werden, sondern auch als ihre Reihenfolge, ihre Zusammenhänge und der Prüfungsprozess der Ergebnisse, die für das überprüfte computergestützte System gelten. Sie müssen Zusammenhänge zwischen Anforderungen, Spezifikationen und Tests herstellen und pflegen. Dieser Zusammenhang ist in der Rückverfolgbarkeitsmatrix dokumentiert, die in den regulierten Bereichen verwendet wird.
Die Rückverfolgbarkeitsmatrix stellt sicher, dass:
- Anforderungen durch das Lösungsdesign erfüllt werden. Mit anderen Worten: Jede Anforderung wird auf die Funktionen, Kontrollen, Konfigurationen oder Designelemente zurückgeführt.
- Anforderungen getestet oder verifiziert werden, um zu demonstrieren, dass das Lösungsdesign die Anforderungen ggf. erfüllt.
Die Vorteile der Rückverfolgbarkeitsmatrix:
- Unterstützt die Überprüfung des Designs
- Hilft bei der Definition des Umfangs der Regressionstests
- Bietet Unterstützung bei Inspektions- oder Überwachungsaktivitäten
- Bietet Unterstützung bei möglichen Änderungen
Plattformqualifikation
Regulierte Branchen müssen Microsoft Power Platform vor der Implementierung von Guides als Infrastruktur qualifizieren. Dafür müssen mindestens die folgenden Aufgaben ausgeführt werden:
Erste Risikobewertung (Bewertung der GxP-Anwendbarkeit)
Lieferantenbewertung (Überprüfung des Lieferanten – kann virtuell, physisch oder postalisch erfolgen)
Qualifikationsplan
Technische Spezifikation des Plattformdesigns
Risikobewertung, zum Beispiel das Risiko, dass Bedienpersonal die falsche Version einer Anleitung zur Verfügung gestellt wird
Tests:
- Installationstests (z. B. Testen, ob die Umgebungen korrekt installiert sind)
- Betriebstests (z. B. Testen, ob die richtigen Benutzer*innen über die richtigen Zugriffsrechte verfügen)
Zusammenfassungsbericht zur Qualifikation
Plattform- oder Betriebshandbuch, Schulungsmaterial
Anwendungsprüfungen
Anwendungen (z. B. Guides und Power Apps), die Geschäftsprozesse in regulierten Branchen unterstützen, müssen überprüft werden. Daher muss Ihre Organisation die folgenden Aufgaben erledigen:
Erste Risikobewertung
Prüfungsplan
Anforderungen der Benutzer*innen
Risikobewertung
Technische Spezifikation für Anwendungsfunktion oder -konfiguration
Tests (Installation Qualification (IQ), Operational Qualification (OQ) und User Annehmenance Testing (UAT)):
- Betriebstests (z. B. Überprüfung einer Funktion)
- UAT
Rückverfolgbarkeitsmatrix
Zusammenfassungsbericht der Überprüfung
Anwendungs- oder Betriebshandbuch, Schulungsmaterial